Sickle cell disease is an inherited blood disorder, it is characterized by the possession of two abnormal hemoglobin, at least one of which is hemoglobin S. There are various types, including HBSS, HBSC and HBS Thalassemia. The types of hemoglobin a person makes in the red blood cells depend upon what hemoglobin genes the person inherits from his or her parents. Like most genes, hemoglobin genes are inherited in two sets, one from each parent.
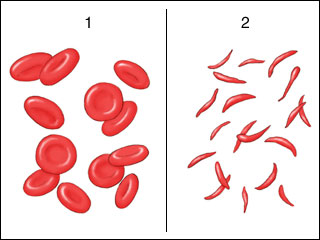
A red blood cell’s primary component is hemoglobin. It facilitates the transport of oxygen from the air in our lungs to every cell in the body. Hemoglobin A is found in regular red blood cells. Hemoglobin A-containing regular red blood cells are soft and rounded, and they can fit through tiny blood vessels (vessels). Usually, red blood cells last for 120 days before being replaced by fresh ones.
Hemoglobin S, a distinct form of hemoglobin A produced by people with sickle cell diseases. Hemoglobin S-containing red blood cells do not survive as long as regular red blood cells do (normally about 16 days). They stiffen and change shape, making it difficult for them to flow through the body’s tiny blood vessels. When sickle-shaped cells block small blood vessels, less blood can reach that part of the body. Parts of the body that don’t receive a normal blood flow eventually become damaged.
What causes sickle cell disease?
- A person will be born with sickle cell disease only if two genes are inherited—one from the mother and one from the father.
- A person who inherits just one gene is healthy and said to be a “carrier” of the disease. A carrier has an increased chance of having a child with sickle cell disease if he or she has a child with another carrier.
What are the symptoms of sickle cell anemia?
Sickle cell anemia symptoms typically start to show about 6 months of age. They can evolve over time and differ from person to person. Signs and symptoms can include:
- joint and bone pain, especially during cold wet seasons
- yellowing of the eyes and skin (jaundice)
- anemia
- difficulty in breathing with or without chest pain
- chronic leg ulcer
- abdominal pain, especially int he splenic area
- delayed growth
- frequent infection
- excessive fatigue or irritability, from anemia
- swelling and pain in hands and feet
How is sickle cell anemia treated?
- Rehydration with intravenous fluids helps red blood cells return to a normal state.
- Folic acid. Folic acid will help prevent severe anemia.
- Pain medication is used to relieve the pain during a sickle crisis
- Daily hydroxyurea reduces the frequency of painful crises and might reduce the need for blood transfusions and hospitalizations. But it can increase the risk of infections. Don’t take the drug if you’re pregnant.
- Blood transfusions improve transport of oxygen and nutrients as needed
- Supplemental oxygen is given through a mask. It makes breathing easier and improves oxygen levels in the blood.
- Vaccinations and antibiotics. These are used to prevent infections.
- Treating underlying or associated infections is an important part of managing the crisis, as the stress of an infection can result in a sickle cell crisis. An infection may also result as a complication of a crisis.
What complications can arise from sickle cell anemia?
- Stroke
- Blindness
- Increased infections
- During pregnancy, intrauterine growth restriction, spontaneous abortion, and pre-eclampsia
- Leg ulcers
- Bone damage
- Early gallstones
- Kidney damage and loss of body water in the urine
- Eye damage
- Multiple organ failure
How Can I Prevent a Crisis?
There’s no sure way, but you can lower your odds:
- Avoid swimming in cold water.
- Dress in warm clothes when it’s cold
- Stay hydrated
- Limit how much alcohol you drink.
- Wash your hands often.
- Manage your stress.
There’s no cure for most people with sickle cell anemia. Treatments can relieve pain and help prevent complications associated with the disease.

Leave a comment